Česká a slovenská psychiatrie

Časopis
Psychiatrické společnosti ČLS JEP
a Psychiatrickej spoločnosti SLS
souborný článek / review article
- AKTUÁLNÍ ČÍSLO
- ARCHIV
- VYHLEDÁVÁNÍ
- PERIODIKUM
- REDAKČNÍ RADA
- PŘEDPLATNÉ
- INZERCE
- AKTUALIZOVANÉ
POKYNY
PRO AUTORY
- ISSN 1212-0383




- © Česká a Slovenská psychiatrie 2025
- © Galén 2025
INZULÍNOVÁ REZISTENCE A NEUROPSYCHIATRICKÁ ONEMOCNĚNÍ
INSULIN RESISTENCE AND NEUROPSYCHIATRIC DISEASES
Miroslav Zeman1, Roman Jirák2, Jiří Raboch2, Marek Vecka1, Aleš Žák1
1IV. Interní klinika
2Psychiatrická klinika 1. lékařské fakulty Univerzity Karlovy v Praze a VFN Praha, U Nemocnice 2, Praha 2
2Psychiatrická klinika 1. lékařské fakulty Univerzity Karlovy v Praze a VFN Praha, U Nemocnice 2, Praha 2
Práce byla podpořena výzkumnými záměry MSM 0021620849 a MSM 0021620820.
SOUHRN
Zeman M, Jirák R, Raboch J, Vecka M, Žák A. Inzulínová rezistence a neuropsychiatrická onemocnění
Rezistence tkání na působení inzulínu se účastní etiopatogeneze diabetů mellitu 2. typu a je také spojena se zvýšenou kardiovaskulární morbiditou i mortalitou. K hlavním patogenetickým mechanismům, které se v působení inzulínové rezistence u těchto onemocnění uplatňují, patří oxidační stres a subklinický zánětlivý proces. V posledních letech bylo zjištěno, že inzulínová rezistence v periferních tkáních i v mozku hraje zřejmě roli také v patogenezi některých neurologických a psychiatrických onemocnění, zejména sporadické formy Alzheimerovy demence, depresivních poruch a schizofrenie, které jsou charakterizovány zvýšeným výskytem diabetů mellitu 2. typu i kardiovaskulárních onemocnění. Inzulínová rezistence a metabolické poruchy s ní spojené zasahují do rozvoje atrofie specifických oblastí v mozku (např. hipokampu), souvisí s tvorbou amyloidových plaků, patologických intraneuronálních klubek i s poruchou struktur zodpovídajících za schopnost paměti a učení. V článku jsou shrnuty mechanismy, kterými se inzulínová rezistence uplatňuje v patogenezi vybraných neuropsychiatrických onemocnění.
Klíčová slova: inzulínová rezistence, oxidační stres, subklinický zánět, Alzheimerova choroba, deprese, schizofrenie, neurodegenerativní onemocnění.
SUMMARY
Zeman M, Jirák R, Raboch J, Vecka M, Žák A. Insulin resistence and neuropsychiatric diseases
Tissue resistance to the insulin effects is taking part in the etiopathogenesis of diabetes mellitus type 2; moreover, it is linked with increased cardiovascular morbitidy and mortality. The principal pathogenetic mechanisms of insulin resistance playing a role in these diseases include oxidative stress and sub clinical inflammatory processes. The findings of the last years revealed the role of peripheral as well as brain insulin resistance in the pathogenesis of some neurological and psychiatrical diseases, particularly the sporadic form of Alzheimer disease, depressive disorders and schizophrenia, all of them being characterized with increased prevalence of diabetes mellitus type 2 and/or cardivascular disease. Insulin resistance and associated metabolic disturbances interfere with the development of specific brain areas (e. g. hippocampus), are linked with the formation of amyloid plaques, pathologic intra-neuronal tangles, and also with the deterioration of the structures responsible for memory and learning processes. The paper reviews the mechanisms, by which insulin resistance influences the pathogenesis of selected neuropsychiatric disorders.
Key words: insulin resistance, oxidative stress, sub clinical inflammation, Alzheimer disease, depression, schizophrenia, neurodegenerative disorders.
ÚVOD
V posledních letech je na celém světě i v České republice zaznamenáván rychle stoupající výskyt kardiovaskulárních chorob (KVO) a diabetů mellitu 2. typu (DM2). Současně stoupá i výskyt neuropsychiatrických onemocnění, jako je demence (Alzheimerova typu i vaskulární) či deprese.80 V etiopatogenezi KVO a DM2 hraje roli inzulínová rezistence (IR),57 která má vztah k některým neuro-psychiatrickým onemocněním, jako jsou např. demence4 nebo depresivní poruchy.45,55,75
INZULÍNOVÁ REZISTENCE - DEFINICE
Inzulínovou resistenci (IR) lze definovat jako soubor metabolických abnormalit a klinických příznaků, provázený sníženou citlivostí tkání na účinek inzulínu. Yalowová s Bersonem definovali IR jako "stav (buňky, tkáně, orgánu nebo organismu), při kterém je k vyvolání kvantitativně normální odpovědi zapotřebí většího než obvyklého množství inzulínu".78 Jednotlivé metabolické dráhy jsou na působení inzulínu různě citlivé. Inzulínová rezistence je spojena se zvýšeným oxidačním stresem, endoteliální dysfunkcí i subklinickým zánětem, které se účastní patogeneze neurodegenerativních změn.65
MOZEK A INZULÍNOVÁ REZISTENCE
Mozek dlouhou dobu nebyl počítán mezi orgány citlivé na působení inzulínu. Později však bylo zjištěno, že v řadě oblastí centrální nervové soustavy (CNS) lze prokázat inzulín i inzulínové receptory Inzulín je vytvářen v beta-buňkách Langerhansových ostrůvků pankreatu a do mozku se dostává přes hematoencefalickou bariéru po vazbě na saturabilní receptorový systém. Inzulín však může být zřejmě syntetizován také v mozku.13 Inzulínové receptory byly identifikovány v hypotalamu, hipokampu, amygdale, v plexus choriodeus i v dalších strukturách.25,70 Většina inzulínových receptorů je lokalizována na neuronech, zatímco gliální buňky mají receptorů jen málo.48 Inzulín se v mozku účastní regulace energetické bilance organismu,22,25,63 přičemž zde hraje úlohu také interakce inzulínu s dalšími faktory, jako např. leptin a serotonin.21 Inzulín reguluje v mozku (za spolupůsobení dalších neuropeptidů) také reprodukční a kognitivní funkce.21,63
Inzulín v mozku, podobně jako v periferních tkáních, po vazbě na receptory stimuluje jejich autofosforylaci a aktivuje signální kaskádu. Je aktivována regulační subjednotka p85 fosfatidyl-inositol-3-kinázy (PI3K) s následnou fosforylací fosfatidyl-inositol-4,5-bisfosfátu (PIP2) na fosfatidyl-inositol-3, 4,5-trisfosfát (PIP3). K PIP3 se váže protein kináza B/AKT a fosfoinositid-dependentní proteinkináza 1 (PDK1). Fosforylovaná PKB vstupuje do buněčného jádra, kde inaktivuje transkripční faktor forkhead box protein Ol (FoxOl).59
V neuronech nucleus arcuatus hypotalamu dochází ke zvýšené expresi proopiomelanokortinu, tvorbě melanokortinu a k poklesu AgRP (agouti-related peptide), s celkově anorexigenním účinkem.23,43 Na snížení příjmu potravy se inzulín podílí v souhře s hormonem leptinem, který využívá stejně jako inzulín signální cestu IRS a PI3K.43 Příjem potravy moduluje také neuropřenašeč serotonin (5-hydroxy tryptamin), jehož aktivita koreluje s inzulínovou senzitivitou.29 Inzulín zvyšuje transport tryptofanu (prekurzoru serotoninu) do mozku40 a serotonin zase zvyšuje citlivost k inzulínu.24
Inzulín v mozku inhibuje aktivaci (firing) neuronů, inhibuje zpětné vychytávání noradrenalinu v mozku, ovlivňuje koncentrace neuropřenašečů (jako noradrenalin nebo acetylcholin), koncentraci mRNA noradrenalinových a dopaminových transportérů v neuronech, moduluje obrat katecholaminů v hypotalamu, stimuluje syntézu fosfoinositolu v hipokampu a podporuje růst neuronů a tvorbu synapsí.75 Inzulín ovlivňuje mechanismy učení a paměti zvýšením exprese NMDA (N-methyl-D-aspartátových) receptorů s ovlivněním dlouhodobé potenciace (long-term potentiation, LTP).64
V posledních letech jsou prokazovány asociace mezi poruchami glukózové homeostázy, zejména DM2, a neurodegenerativními onemocněními. U nemocných s DM2 se zjišťují poruchy paměťových funkcí.14 U osob s chronickou hyperinzulinémií byla i v případě normální hladiny glykémie porušena slovní paměť.73
Proto je dnes věnována pozornost souvislostem mezi hyperinzulinémií, inzulínovou rezistencí a patofyziologií Alzheimerovy nemoci, ale i dalších neuropsychiatrických onemocnění.
NEUROPSYCHIATRICKÁ ONEMOCNĚNÍ SPOJENÁ S INZULÍNOVOU REZISTENCÍ
Alzheimerova choroba
Alzheimerova nemoc (AD) je nejčastější příčinou demence a její prevalence stále stoupá na celém světě, což je dáno především celosvětovým stárnutím populace - věk je hlavním rizikovým faktorem AD.18 Alzheimerova nemoc je heterogenní onemocnění, charakterizované ukládáním amyloidu beta (Aβ) v mozku a hromaděním degenerovaného proteinu tau v neuronech.33 Neurotoxicky působí zejména oligomery Aβ (označované také jako ADDLs; Aβ derived diffusible ligands), jsou vlastními neurotoxiny ADDLs působí jako patogenní ligandy na synapsích neuronů; důsledkem je inhibice LTP, oxidační stres, poškození synapsí a také hyperfosforylace tau.9
Rozlišují se dvě základní skupiny AD: (1) familiární forma, zřejmě geneticky podmíněná, a (2) pozdní (sporadická) forma, v jejíž patogenezi hrají úlohu faktory, jako je věk, vysoký příjem kalorií a nasycených tuků ve stravě, kouření cigaret, hyperhomocysteinémie nebo diabetes mellitus.38 Sporadická forma představuje nyní asi 90 % všech případů AD. Podle řady autorů má klíčovou roli v patogenezi pozdní formy AD hyperinzulinémie, spojená s porušenou signální funkcí inzulínu centrálně, v CNS.67 Inzulín kolující v periferní krvi prostupuje hematoencefalickou bariéru a po vazbě na inzulínové receptory ovlivňuje několik mechanismů, spojených s patogenezí AD. Inzulín zvyšuje degradaci Aβ v neuronech;58 aktivace inzulínového receptoru v mozku vede ke zvýšené expresi inzulin-degradujícího enzymu (IDE), který vedle štěpení molekuly inzulínu štěpí také Aβ a intracelulární doménu fragmentu APP (intracellular domain fragment APP). Inzulín kompetuje sApo IDE, a tak hyperinzulinémie může přispívat ke zvýšené zátěži Aβ. Inzulínová signalizace v mozku vede také k inhibici aktivity enzymu glykogensyntázy kinázy 3 beta (GSK3 ) a tím ke zvýšení aktivity GSK3β, která zvyšuje fosforylaci proteinu tau a tak tvorbu patologických intra-neuronálních neurofibrilárních klubek (neurofibrillar tangles, NT). Porušená inzulínová signalizace je také spojena se zvýšením aktivity cdk5 (cyklin dependentní kinázy), což je enzym, který se rovněž podílí na hyperfosforylaci tau.12
Na clearanci Aβ se podílí také interakce inzulínu se systémem IGF (Insulin-like growth factor I, somatomedin-C A). IGF je polypeptidový hormon strukturálně podobný proinzulinu, vytvářený v játrech a fibroblastech. Inzulín působí uvolňování intracelulárního Aβ, který je pak navázán na proteinový nosič, indukovaný IGF a transportován ven z mozku.20 Tento proces je inhibován zánětlivým cytokinem TNFα (tumor necrosis factor alfa).
V patogenezi Alzheimerovy choroby se uplatňují také tzv. konečné produkty pokročilé glykace, AGEs (advanced glycation end products), vznikající neenzymatickou vazbou karbonylových sloučenin včetně redukujících cukrů na aminoskupiny proteinů. Jejich koncentrace je zvýšena u nemocných s diabetem mellitem, ale i u osob s porušenou glukózovou tolerancí (PGT), aterosklerózou či renální insuficiencí. AGEs jsou ukazatele karbonylového stresu, které se hromadí v důsledku zvýšené hladiny sacharidů (glukóza, fruktóza, deoxyglukóza) a reaktivních dikarbonylových sloučenin (např. glyoxal, metylglyoxal a 3-deoxyglukoson).53
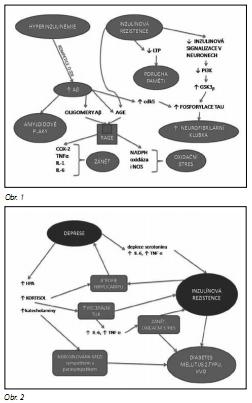
Porucha glukózového metabolismu vede k vzestupu intracelulárních reaktivních karbonylových sloučenin (např. metylglyoxal). Přechodné kovy, jako je železo nebo měď, které se hromadí v amyloidových plácích, urychlují oxidaci glykovaných proteinů a působí tak zvýšení hladiny glykoxidačních produktů, a zvyšují i agregaci Aβ. Negativní roli hraje také pokles hladin látek, snižujících glykan, oxidaci a tvorbu AGE, jako je anserin nebo karnosin.27 Zvýšená tvorba AGE může být také způsobena poklesem aktivity enzymu glyceral-dehyd-3-fosfát dehydrogenázy (GAPDH), která vede ke zvýšení hladiny glukózy a intermediátů glykolýzy s následným zvýšením aktivity polyolové a hexosaminové cesty, k aktivaci protein kinázy C a zvýšené tvorbě AGE.3
AGEs jsou ligandy pro receptory (RAGE, receptor for advanced glycation endproducts). Po jejich vazbě na RAGE dochází mimo jiné k aktivaci jaderného transkripčního faktoru kappa B (NFkB) s následnou indukcí exprese cytokinů či enzymů, jako je inducibilní syntáza oxidu dusnatého (iNOS), COX-2 (cyklooxygenáza-2) či TNFα a k zánětlivé reakci. Mezi ligandy RAGE patří také Aβ.54 U AD je popisována zvýšená exprese RAGE, RAGE jsou spolu s Aβ vychytávány do astrocytů a degradovány v lyzozomech. V mozku byla exprese RAGE popsána na neuronech a mikrogliích hipokampu, entorhinálního kortexu a horního čelního laloku jak u nemocných s AD, tak i u kontrolních osob bez demence, přičemž u AD byl prokázán zvýšený počet RAGE na mikrogliích.37
Dvě sekreční isoformy RAGE (spliced varianty esRAGE a sRAGE) postrádají transmembránovou doménu, a tak cirkulují v plasmě. sRAGE může kompeticí s RAGE na buněčných površích odstraňovat nebo neutralizovat ligandy RAGE. Vyšší hladiny sRAGE jsou spojeny se sníženým rizikem chorob spojených s chronickým zánětem nebo oxidačním stresem. sRAGE jsou také možným markerem rozvoje neurodegenerativních onemocnění CNS, např. snížené hodnoty sRAGE byly nalezeny u AD.15
Vztahy mezi stavy, spojenými s hyperinzulinémií či inzulínovou rezistencí (PGT či DM2) a výskytem AD, byly prokázány v klinických studiích. Diabetes je spojen se zvýšeným rizikem demence.14,46 Riziko Alzheimerovy i vaskulární demence je zvyšováno nejen přítomností DM2, ale i samotnou IR.38,46-48 Nemocní s časnými stadii AD jsou ve srovnání se zdravými stejně starými osobami a stejnou fyzickou aktivitou charakterizováni menší citlivostí k působení inzulínu.8
V terapii Alzheimerovy choroby jsou zkoušena farmaka, která rezistenci vůči inzulínu snižují, především antidiabetika ze skupiny glitazonů,76 která stimulují receptor PPAR gamma (peroxisome proliferator activated receptor) a snižují toxicitu Aβ i zánětlivou reakci, která se rozvíjí v okolí extracelulárních kortikálních akumulací Aβ - plak. U kmene transgenních myší byl prokázán efekt glitazonového antidiabetika pioglitazonu na snížení gliálního zánětu a hladin mozkového beta-amyloidu Aβ1-42.26 I když stále probíhají klinické studie, byl prokázán pozitivní efekt glitazonových antidiabetik (rosiglitazon, pioglitazon) především na časná stadia Alzheimerovy choroby. Bylo prokázáno zlepšení kognitivních funkcí, je předpokládán neuroprotektivní efekt. Výsledky studií vedly k tomu, že americká léková agentura FDA (Food and Drug Administration) prohlásila tato léčiva za potenciálně užitečná v léčbě Alzheimerovy choroby.35 V terapii Alzheimerovy choroby jsou v klinických studiích testovány statiny Předpokládá se, že zvyšují aktivitu alfa-sekretáz, štěpících APP na krátké fragmenty, které nejsou schopny vytvářet Aβ.36 Rovněž jsou ověřovány inhibitory GSK3β, jejíž nadměrná aktivace je pozorována u IR a která spouští hyperfosforylaci tau, tvorbu neurofibrilárních klubek a zánik neuronu. Zkoušeny jsou např. soli lithia i další látky. Mechanismy, kterými se IR uplatňuje v patogenezi sporadické formy Alzheimerovy demence, jsou znázorněny na obr. 1.
Depresivní porucha (DP)
Mechanismus uplatnění IR u deprese není přesně znám, předpokládá se např. porucha utilizace glukózy v mozku v důsledku IR s následnými změnami neuronů limbického systému, význam má zřejmě také subklinický zánět a oxidační stres, spojený s IR. Depresivní porucha je v současnosti pokládána za určitý typ neurodegenerativního onemocnění.
U nemocných s DP je pozorována odlišná odpověď na stresující vlivy, kde hlavní roli hraje centrální nervový systém.50 Zpracování stresových podnětů v mozku vede k odpovědím autonomního systému, osy hypothalamus-hypofýza-nadledvina i systému oběhového a imunitního. Poruchy těchto funkcí jsou pozorovány u MetS (metabolický syndrom) i u depresí a vztahy mezi IR a depresí byly opakovaně publikovány45 U nemocných s DP je často zjišťována nadprodukce kortikotropního hormonu (corticotropin-releasing factor, CRF), zvýšená hladina kortisolu, porucha suprese jeho sekrece při dexametazonovém supresním testu a dysfunkce osy hypothalamus-hypofýza-nadledvina (HPA) s poruchou cirkadiánního rytmu (aktivace ve večerních hodinách).23 Dysfunkce osy HPA, vzestup sekrece glukokortikoidů a aktivace sympatického nervového systému vede k rezistenci na působení inzulínu. Inzulínová rezistence zvyšuje kortizolovou neurotoxicitu v hipokampu (klíčové oblasti regulace aktivity osy HPA), což může stát v pozadí endokrinního ovlivnění nálady a kognitivních funkcí.55 Hypotézu o vztahu IR a deprese podporují nálezy zvýšeného výskytu deprese u pacientek se syndromem polycystických ovarií, který je charakterizován androgenní hypersekrecí, IR a chronickou anovulací.56 U žen s depresivní poruchou byly prokázány některé rysy MetS (zvýšená glykémie, triglyceridémie, hodnota indexu inzulínové rezistence HOMA (homeostasis model assessment) bez závislosti na léčbě.81
Podstata vztahů mezi depresí a IR, případně mezi léčbou antidepresivy a IR, je v současné době předmětem výzkumu. Deprese může několika způsoby přispívat k rozvoji IR, případně DM2. Hyperkortisolémie je spojena se zvýšenou odpovědí inzulínu a glukózy na glukózovou zátěž.77 Glukokortikoidy působí zvýšené ukládání tuku v adipocytech, zejména ve viscerální oblasti, kde je vyšší hustota glukokortikoidových receptorů než v periferních adipocytech, a současně působí rezistenci na účinky inzulínu. Hyperkortisolémie tak přispívá k rozvoji dvou základních článků MetS: abdominální obezity a IR. Deplece serotoninu v mozku může také přispívat k rozvoji IR29 a léčba SSRI (selective serotonin reuptake inhibitors) preparátem fiuoxetinem vedla u obézních nemocných s DM2 k úpravě inzulínové citlivosti nezávisle na účinku fluoxetinu na hmotnost.39 Zvýšená prozánětlivá aktivita, pozorovaná u deprese, je dalším mechanismem potenciálně přispívajícím k rozvoji IR. U deprese je popisováno zvýšení hladiny IL-1 (interleukin-1) a TNFα, které interferují s inzulínovou signalizací.30 Inzulínovou rezistenci v játrech a tukové tkáni (ale ne v kosterním svalu) působí IL-6,41 jehož hladina byla v rozsáhlé prospektivní studii spolu s koncentrací C-reaktivního proteinu prediktorem vzniku DM2.52 Vztahy mezi depresí, inzulínovou rezistencí a klinickými důsledky jsou znázorněny na obr. 2. Podle některých autorů je deprese rizikovým faktorem Alzheimerovy choroby (AD),7 někteří ji považují za prekurzor tohoto onemocnění.32 Jsou sledovány případy rezistentních, provleklých depresivních poruch, které přecházejí do obrazu demence.
Schizofrenie
Onemocnění schizofrenií je provázeno významně zvýšenou mortalitou na KVO i celkovou mortalitou.10 Nemocní se schizofrenií trpí významně častěji DM2.5 V pozadí těchto jevů stojí zřejmě více faktorů. Nemocní s těžkými psychotickými onemocněními žijí obvykle nezdravým životním stylem (nedostatek fyzické aktivity, kouření cigaret, strava s nadbytkem sacharidů a tuků).11,47 U pacientů se schizofrenií je často nacházen MetS (současný výskyt obezity, arteriální hypertenze, dyslipidémie a poruch homeostázy glukózy a dalších rizikových faktorů KVO). Rozvoj obezity také podporuje antipsychotická léčba; zejména podávání atypických antipsychotik (clozapin, olanzapin) je spojeno s hmotnostním přírůstkem, zvýšením hladin plazmatických lipidů a se zvýšením rizika DM2.10 Hmotnostní nárůst bývá nejvyšší po podávání clozapinu a olanzapinu, v menší míře po risperidonu a quetiapinu a nejméně po aripiprazolu a ziprasidonu, hladiny lipidů jsou nejméně ovlivňovány ziprasidonem a amisulpridem.69 Antipsychotika ovšem také v různé míře nepříznivě ovlivňují homeostázu glukózy nezávisle na nárůstu hmotnosti přímou interferencí s účinky inzulínu s následnou inzulinorezistencí.69
Schizofrenií nepostižení prvostupňoví příbuzní schizofreniků mají významně vyšší výskyt DM2 (cca 19-30 %).42 Poruchy glukózové homeostázy byly u duševních poruch pozorovány ještě před zavedením atypických antipsychotik do praxe a více než 15 % dosud neléčených ("drug naive") pacientů se schizofrenií má zvýšenou lačnou glykémii, inzulinémii, kortisolémii a nižší inzulínovou senzitivitu, měřenou pomocí HOMA.59,66 U dosud neléčených nemocných se schizofrenií byla prokázána IR v játrech, která nesouvisela s intraabdominálním tukem ani s dalšími determinantami IR.71
Bipolární afektivní porucha (BAP)
Inzulínová rezistence má podle několika studií vztah rovněž k patogenezi bipolární afektivní poruchy. V několika studiích byly sledovány vztahy mezi výskytem BAP a MetS (u kterého je inzulínová rezistence spolu s abdominální obezitou považována za klíčovou komponentu).57 V řadě studií byla u nemocných s BAP zjištěna mírně, ale statisticky významně vyšší prevalence MetS ve srovnání s běžnou populací.16,19,79 Na výskytu MetS nemocných s BAP se může podílet i léčba tzv. atypickými antipsychotiky (antipsychotiky druhé generace).69
Huntingtonova choroba (HD)
Jde o autozomálně dominantně dědičné neurodegenerativní onemocnění, postihující asi jednoho z 10 000 lidí. Příčinou je nestabilní expanze trinukleotidové repetice CAG v exonu 1 v genu pro bílkovinu huntingtin, nacházejícím se na krátkém raménku chromozomu 4. V důsledku zmnožení polyglutaminu v molekule huntingtinu dochází k jeho agregaci a tvorbě neuronálních inkluzních tělísek.28 Patologicko-anatomicky je onemocnění charakterizováno degenerací neuronů ve striatu s výraznou atrofií putamen a nucleus caudatus. Huntingtonova nemoc je další neurodegenerativní poruchou, u které je pozorován zvýšený výskyt IR a DM2. V jedné studii u 620 nemocných s HD byla ve srovnání se stejně starými jedinci běžné populace zjištěna významně vyšší prevalence DM2 (10,5 %).17 Podle některých autorů by mohl být v pozadí jevu pokles sekrece inzulínu u nemocných s HD.51 U transgenních myší s HD byla v buňkách Langerhansových ostrůvků pankreasu prokázána snížená exprese inzulínu, ale i somatostatinu a glukagonu a byly zjištěny intranukleární inkluze, hromadící se s věkem, které korelovaly s postupným poklesem glukózové tolerance.1
U normoglykemických nemocných s HD byly ve srovnání se zdravými osobami zjištěny vyšší hodnoty IR vyjádřené HOMA indexem a nižší hodnoty indexu inzulínové citlivosti. Současně při vyšetření časné fáze inzulínové sekrece měli nemocní s HD nižší akutní inzulínovou odpověď a nižší inzulinogenní index.34 Patogenetický význam u HD má také oxidační stres a mitochondriální dysfunkce, které se uplatňují na jedné straně i u dalších neurodegenerativních chorob,65 a na druhé straně mají význam též u diabetů mellitu.6
Parkinsonova choroba (PN)
Základním patologicko-anatomickým rysem PN je ztráta dopaminergních neuronů v substantia nigra - pars compacta, která postihuje přímé a nepřímé inhibiční cesty do globus pallidus a substantia nigra reticulata. V neuro-patologických studiích byl u nemocných s PN prokázán deficit m-RNA tyrosinové hydroxylázy, klíčového enzymu pro syntézu dopaminu.68 U Parkinsonovy nemoci (PN) se vyskytuje až v 50-80 % případů PGT.61 Podávání některých antiparkinsonik (např. levodopa) může vést ke zvyšování hladiny glykémie a inzulinémie. Jiná farmaka, užívaná v léčbě PN, však mohou naopak inzulínovou senzitivitu zvyšovat. Nálezy klinických studií ukazují, že IR může manifestaci PN předcházet, podobně jako je tomu u deprese či schizofrenie. U nově zjištěných případů s PN byl prokázán pokles inzulínové senzitivity.72 Byl studován vztah PN k DM2, nálezy však nejsou jednotné. Některé studie naznačují asociace mezi DM2 a PN,31 jiné práce tyto nálezy nepotvrzují.62
LITERATURA
- 1. Andreassen OA, Dedeoglu A, Stanojevic V et al. Huntingdon's disease of the endocrine pancreas: insulin deficiency and diabetes mellitus due to impaired insulin gene expression. Neurobiol Dis 2002;11: 410-424.
- 2. Ballatore C, Lee VM, Trojanowski JQ. Tau-mediated neuro degeneration in Alzheimer's disease and related disorders. Nat Rev Neurosci 2007; 8: 663-672.
- 3. Blatník M, Thorpe SR, Baynes JW. Succination of Proteins by Fumarate: Mechanism of Inactivation of Glyce-raldehyde-3-Phosphate Dehydrogenase in Diabetes. Ann N Y Acad Sci 2008; 1126: 272-275.
- 4. Carantoni M, Zuliania G, Munarib MR, D'Eliaa K, Palmieria E, Fellina R. Alzheimer Disease and Vascular Dementia: Relationships with Fasting Glucose and Insulin Levels. Dement Geriatr Cogn Disord 2000; 11: 176-180.
- 5. Casey DE. Metabolic issues and cardiovascular disease in patients with psychiatric disorders. Am J Med 2005; 118: 15S-22S.
- 6. Ceriello A, Motz E. Is oxidative stress the pathogenic mechanism underlying insulin resistance, diabetes, and cardiovascular disease? The common soil hypothesis revisited. Arterioscler Thromb Vase Biol 2004; 24: 816-823.
- 7. Cooper B, Holmes C. Previous psychiatric history as a risk factor for late-life dementia: a population-based case-control study. Age Ageing 1998; 27: 181-188.
- 8. Craft S, Newcomer J, Kanne S et al. Memory improvement following induced hyperinsulinemia in Alzheimer's disease. Neurobiol Aging 1996; 17: 123-30.
- 9. De Felice FG, Vieira MNN, Bomfim TR, Decker H, Velasco PT, Lambert MP, Viola KL, Zhao W-Q, Ferreira ST, Klein WL. Protection of synapses against Alzheimer's linked toxins: insulin signaling prevents the pathogenic binding of Ap oligomers. PNAS 2009, 106: 1971-1976.
- 10. De Hert M, Dekker JM, Wood D, Kahl KG, Holt RI, Moller HJ. Cardiovascular disease and diabetes in people with severe mental illness position statement from the European Psychiatric Association (EPA), supported by the European Association for the Study of Diabetes (EASD) and the European Society of Cardiology (ESC). Eur Psychiatry 2009; 24: 412-424.
- 11. De Hert M, Schreurs V, Vancampfort D, Van Winkel R. Metabolic syndrome in people with schizophrenia. World Psychiatry 2009; 8: 15-22.
- 12. de la Monte SM, Wands JR. Review of insulin and insulin-like growth factor expression, signaling, and malfunction in the central nervous system: Relevance to Alzheimer's disease. J Alzheimers Dis 2005; 7: 45-61.
- 13. Devaskar SU, Giddings SJ, Rajakumar PA, Carnaghi LR, Menon RK, Zahm DS. Insulin gene expression and insulin synthesis in mammalian neuronal eels. J Biol Chem 1994; 269: 8445-8454.
- 14. Elias PK, Elias MF, DAgostino RB et al. NIDDM and blood pressure as risk factors for poor cognitive performance: The Framingham Study. Diabetes Care 1997, 20: 1388-1395.
- 15. Emanuele E, DAngelo A, Tomaino C et al. Circulating levels of soluble receptor for advanced glycation end products in Alzheimer disease and vascular dementia. Arch Neurol 2005; 62(11): 1734-1736.
- 16. Fagiolini A, Frank E, Scott JA, Turkin S, Kupfer DJ. Metabolic syndrome in bipolar disorder: findings from the Bipolar Disorder Center for Pennsylvanians. Bipolar Disord. 2005; 7: 424-430.
- 17. Farrer LA. Diabetes mellitus in Huntington's disease. Clin Genet 1985; 27(1): 62-67.
- 18. Ferri CP, Prince M, Brayne C et al. Global prevalence of dementia: a Delphi consensus study. Lancet 2005; 366: 2112-2117.
- 19. Garcia-Portilla MP, Saiz PA, Benabarre A et al. The prevalence of metabolic syndrome in patients with bipolar disorder. J Affect Disord 2008; 106: 197-201.
- 20. Gasparini L, Xu H. Potential roles of insulin and IGF-1 in Alzheimer's disease. Trends Neurosci. 2003; 26: 404-406.
- 21. Gerozissis K. Brain insulin and feeding: a bi-directional communication. Eur J Pharmacol 2004; 490 (1-3): 59-70.
- 22. Gerozissis K. Brain insulin: Regulation, Mechanisms of action and Functions. Cell Mol Neurobiol 2003; 23: 1-24.
- 23. Gillespie CF, Nemeroff CB. Hypercortisolemia and depression. Psychosom Med 2005; 67 Suppl 1: S26-28.
- 24. Goodnick PJ, Henry JH, Buki VMV. Treatment of depression in patients with diabetes mellitus. J Clin Psychiatry 1995; 56: 128-136.
- 25. Havránkova J, Roth J, Brownstein M. Insulin receptors are widely distributed in the central nervous system of the rat. Nature 1978, 272: 827-829.
- 26. Heneka MT, Sastre M, Dumitrescu-Ozimek L, Hanke A, Dewachter I, Kuipieri C, O'Banion K, Klockgether T, Van Leuven F, Landreth GE. Acute treatment with PPARgamma agonist pioglitazone and ibuprofen reduces glial inflammation and Abetal-42 levels in APPV7171 transgenic mice. Brain 2005; 128: 1442-53.
- 27. Hipkiss AR. Carnosine, diabetes and Alzheimer's disease. Expert Rev Neurother 2009; 9: 583-585.
- 28. Hoffner G, Souěs S, Djian P. Aggregation of expanded huntingtin in the brains of patients with Huntington disease. Prion 2007; 1: 26-31.
- 29. Horáček J, Kuzmiaková M, Hoschl C, Anděl M, Bahbonh R. The relationship between central serotonergic activity and insulin sensitivity in healthy volunteers. Psychoneuroendocrinology 1999; 24: 785-797.
- 30. Hotamisligil GS. Inflammatory pathways and insulin action. Int J Obes2003; 27:S53-S55.
- 31. Hu G, Jousilahti P, Bidel S et al. Type 2 diabetes and the risk of Parkinson's disease. Diabetes Care 2007; 30: 842-847.
- 32. Chen P, Ganguli M, Mulsant BH, DeKosky ST. The temporal relationship between depressive symptoms and dementia: a community based prospective study. Arch Gen Psychiatry 1999; 56: 261-266.
- 33. Jirák R, Koukolik F Demence. Praha: Galén 2004.
- 34. Lalič NM, Maric J, Světel M et al. Glucose Homeostasis in Huntington Disease. Abnormalities in Insulin Sensitivity and Early-Phase Insulin Secretion. Arch Neurol 2008; 65 (4): 476-480.
- 35. Landreth G. Therapeutic use of agonists of the nuclear receptor PPARgamma in Alzheimer's disease. Curr Alzheimer Res 2007; 4: 159-164.
- 36. Lichtenthaler SF, Haass C. Amyloid at the cutting edge: activation of alpha-secretase prevents amyloidogenesis in an Alzheimer disease mouse model. J Clin Invest 2004; 113: 1384-1387.
- 37. Lue L-F, Walker DG, Brachova L, Beach TG, Rogers J, Schmidt AM, Stern DM, Yan SD. Involvement of Microglial Receptor for Advanced Glycation Endproducts (RAGE) in Alzheimer's Disease: Identification of a Cellular Activation Mechanism Experimental Neurology 2001; 171: 29-45.
- 38. Luchsinger JA, Mayeux R. Adiposity and Alzheimer's disease. Curr Alzheimer Res 2007, 4: 127-134.
- 39. Maheux P, Ducros F, Bourque J, Garon J, Chiasson JL. Fluoxetine improves insulin sensitivity in obese patients with non-insulin-dependent diabetes mellitus independently of weight loss. Int J Obes Relat Metab Disord 1997; 2: 97-102.
- 40. Malone KM, Thase ME, DeMeo M et al. Fenfluramine challenge test as a predictor of outcome in major depression. Psychopharmacol Bull 1993; 229: 155-161.
- 41. Montecucco F, Steffens S, Mach E Insulin Resistance: A Proinflamma-tory State Mediated by Lipid-Induced Signaling Dysfunction and Involved in Atherosclerotic Plaque Instability. Mediators Inflamm 2008; 2008: 767623.
- 42. Mukherjee S, Schnur DB, Reddy R Family history of type 2 diabetes in schizophrenic patients. Lancet 1989; 1: 495.
- 43. Niswender KD, Morrison CD, Clegg DJ, Olson R, Baskin DG, Myers MG Jr, Seeley RJ, Schwartz MW. Insulin activation of phosphatidylinositol 3-kinase in the hypothalamic arcuate nucleus: a key mediator of insulin-induced anorexia. Diabetes, 2003; 52: 227-231.
- 44. Okamura F, Tashiro A, Utumi A et al. Insulin resistance in patients with depression and its changes during the clinical course of depression: minimal model analysis. Metabolism 2000; 49: 1255-1260.
- 46. Ott A, Stolk RP, van Harskamp F, Pols HA, Hofman A, Breteler MM. Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology. 1999; 53: 1937-1942.
- 47. OudMJT, Meyboom-de Jong B. Somatic diseases in patients with schizophrenia in general practice: their prevalence and health care, BMC Family Practice 2009, 10: 32.
- 48. Park CR. Cognitive effects of insulin in the central nervous system. Neurosci Biobehav Rev 2001; 25(4): 311-323.
- 49. Peila R, Rodriguez BL, Launer LJ. Honolulu-Asia Aging Study. Type 2 diabetes, APOE gene, and the risk for dementia and related pathologies: The Honolulu-Asia Aging Study. Diabetes 2002; 51: 1256-1262.
- 50. Pittenger C, Dumán RS. Stress, Depression, and Neuroplasticity: A Convergence of Mechanisms. Neu-ropsychopharmacology Reviews 2008; 33: 88-109.
- 51. Podolsky S, Leopold NA. Abnormal glucose tolerance and arginine tolerance tests in Huntington's disease. Gerontology 1977; 23 (1): 55-63.
- 52. Pradhan AD, Manson JE, Rifai N, Buring JE, Ridker PM. C-Reactive Protein, Interleukin 6, and Risk of Developing Type 2 Diabetes Mellitus. JAMA 2001; 286: 327-334.
- 53. Rabbani N, Thornalley PJ. Dicarbonyls linked to damage in the powerhouse: glycation of mitochondrial proteins and oxidative stress. Biochem Soc Trans. 2008; 36: 1045-1050.
- 54. Ramasamy R, Vannucci SJ, Yan SS et al. Advanced glycation end products and RAGE: a common thread in aging, diabetes, neurodegeneration, and inflammation. Glycobiology 2005; 15, 16R-28R.
- 55. Rasgon N, Jarvik L. Insulin Resistance, Affective Disorders, and Alzheimer's Disease: Review and Hypothesis. J Gerontol 2004; 59A: 178-183.
- 56. Rasgon NL, Rao RC, Hwang S et al. Depression in women with polycystic ovary syndrome: clinical and biochemical correlates. J Affect Disord 2003; 74: 299-304.
- 57. Reaven, GM. Metabolic syndrome. Pathophysiology and implications for management of cardiovascular disease. Circulation 2002; 106: 286-288.
- 58. Reger MA, Watson GS, Green PS et al. Intranasal insulin improves cognition and modulates beta-amyloid in early AD. Neurology 2008; 70: 440-448.
- 59. Ryan MC, Collins P, Thakore JH. Impaired fasting glucose tolerance in first-episode, drug-naive patients with schizophrenia. Am J Psychiatry 2003, 160 (2): 284-289.
- 60. Salih DAM, Brunet A. FoxO transcription factors in the maintenance of cellular homeostasis during aging. Curr Opin Cell Biol 2008; 20: 126-136.
- 61. Sandyk R. The relationship between diabetes mellitus and Parkinson's disease. Int J Neurosci 1993; 69 (1-4): 125-130.
- 62. Scigliano G, Musicco M, Soliveri P et al.: Reduced risk factors for vascular disorders in Parkinson disease patients: a case control study. Stroke 2006; 37: 1184-1188.
- 63. Schwartz MW, Woods SC, Porte Jr D, Seeley RJ, Baskin DC. Central nervous system control of food intake. Nature 2000; 404: 661-671.
- 64. Skeberdis VA, Lan J, Zheng X, Zukin RS, Bennett MV. Insulin promotes rapid delivery of N-methyl-D-aspartate receptors to the cell surface by exocytosis. Proc Natl Acad Sci U.S.A. 2001;98:3561-3566.
- 65. Skovronsky DM, Lee VM, Trojanowski JQ. Neurodegenerative Diseases: New Concepts of Pathogenesis and Their Therapeutic Implications. Annu Rev Pathol Mech Dis 2006; 1: 151-70.
- 66. Spelman LM, Walsh PI, Sharifi N, Collins P, Thakore JH. Impaired glucose tolerance in first-episode drug-naive patients with schizophrenia. Diabet Med 2007; 24: 481-485.
- 67. Strachan MWJ. Insulin and cognitive function in humans: experimental data and therapeutic considerations. Biochem Soc Transact 2005; 13: 1037-1040.
- 68. Takahashi M, Yamada T, Tooyama I et al. Insulin receptor mRNA in the substantia nigra in Parkinsons disease. Neurosci Lett 1996; 204: 201-204.
- 69. Tschoner A, Engl J, Laimer M et al. Metabolic side-effects of antipsychotic medication. Int. J Clin Pract 2007; 61: 1356-1370.
- 70. Unger JW, Livingston JN, Moss AM. Insulin receptors in the central nervous system: localization.signalling mechanisms and functional aspects. Prog Neurobiol 1991; 36: 343-362.
- 71. van Nimwegen LJ, Storosum JG, Blu-mer RM, Allick G, Venema HW, de Haan L, Becker H, van Amelsvoort T, Ackermans MT, Fliers E, Serlie MJ, Sauerwein HP. Hepatic insulin resistance in antipsychotic naive schizophrenic patients: stable isotope studies of glucose metabolism. J Clin Endocrinol Metab 2008; 93: 572-577.
- 72. Van Woert MH, Mueller PS, Ambani LM, Rathey U. Abnormal insulin secretion in Parkinson's disease before and during L-dihydroxyphenylalanine (L-dopa) therapy. J Endocrinol 1973; 59 (3): 523-534.
- 73. Vanhanen M, Koivisto K, Kuusisto J et al. Cognitive function in an elderly population with persistent impaired glucose tolerance. Diabetes Care 1998; 21: 398-402.
- 74. Watson GS, Craft S. Modulation of memory by insulin and glucose: neuropsychological observations in Alzheimer's disease. Eur J Pharmacol 2004; 490 (1-3): 97-113.
- 75. Watson GS, Craft S. Insulin resistance, inflammation, and cognition in Alzheimer's Disease. Lessons for multiple sclerosis. J Neurol Sci 2006; 245: 21-33.
- 76. Watson GS, Cholerton BA, Reger MA et al. Preserved cognition in patients with early Alzheimer disease and amnestic mild cognitive impairment during treatment with rosiglitazone: a preliminary study. Am J Geriatr Psychiatry 2005; 13: 950-958.
- 77. Weber B, Schweiger U, Deuschle M, Heuser I. Major depression and impaired glucose tolerance. Exp Clin Endocrinol Diab 2000; 108: 187-190.
- 78. Yalow RS, Berson SA. Plasma insulin concentrations in nondiabetic and early diabetic subjects. Determination by a new sensitive immuno assay technique. Diabetes 1960; 9: 254-260.
- 79. Yumru M, Savas HA, Kurt E et al. Atypical antipsychotics related metabolic syndrome in bipolar patients. J Affect Disord 2007; 98, 247-252.
- 80. Zdravotnická ročenka České republiky 2007. ÚZIS: Praha 2008.
- 81. Zeman M, Jirák R, Zák A et al. Rysy metabolického syndromu u nemocných s depresivní poruchou. Čas Lék čes 2009; 148: 309-314.