Česká a slovenská psychiatrie

Časopis
Psychiatrické společnosti ČLS JEP
a Psychiatrickej spoločnosti SLS
souborný článek / review article
- AKTUÁLNÍ ČÍSLO
- ARCHIV
- VYHLEDÁVÁNÍ
- PERIODIKUM
- REDAKČNÍ RADA
- PŘEDPLATNÉ
- INZERCE
- AKTUALIZOVANÉ
POKYNY
PRO AUTORY
- ISSN 1212-0383




- © Česká a Slovenská psychiatrie 2025
- © Galén 2025
PSYCHICKÉ PROJEVY NIEMANNOVY - PICKOVY CHOROBY TYPU C
PSYCHIATRIE MANIFESTATIONS OF NIEMANN-PICK DISEASE TYPE C
Jakub Albrecht1, Martin Anders1
1 Psychiatrická klinika 1. LF UK a VFN, Praha
SOUHRN
Albrecht J, Anders M. Psychické projevy Niemannovy-Pickovy choroby typu C
Niemannova-Pickova choroba typu C je autosomálně recesivně dědičné střídavé onemocnění vyskytující se panetnicky s incidencí cca 1 : 150 000. Genetická mutace genu NPC1 nebo NPC2 má za následek poruchu transportu a esterifikace cholesterolu uvnitř buňky. V lyzosomech se hromadí neesterifikovaný cholesterol, sfingomyelin, fosfolipidy a glykosfingolipidy Klinická manifestace Niemannovy-Pickovy choroby typu C je závislá na věku a projevy onemocnění zahrnují kromě hepatosplenomegalie mentální retardaci, poruchy chůze, kataplexii a ataxii doprovázené parézou vertikálního pohledu. Onemocnění nebývá pro variabilitu klinických projevů rozpoznáno, je nesprávně diagnostikováno nebo je zjištěno opožděně a postiženým bývá podávána nevhodná léčba. Plnému rozvoji onemocnění mohou předcházet psychotické příznaky, halucinace nebo agresivní chování. Vzhledem k tomu, že existuje léčba, je nezbytné zvýšit povědomí o této chorobě a její diagnostice tak, aby mohla být včas zahájena její adekvátní terapie.
Klíčová slova: membrány, glykosfingolipidy, neuropsychiatrická onemocnění, gen NPC1, gen NPC2, Niemannova-Pickova choroba typu C
SUMMARY
Albrecht J, Anders M. Psychiatrie Manifestations of Niemann-Pick Disease type C
Niemann-Pick disease type C is a rare and fatal inherited autosomal recessive disease caused by mutations in either the NPC1 or NPC2 genes and leads to the inhibition of unesterified cholesterol glycosphingolipids transport and intracellular accumulation in the brain, liver and spleen. Disease affects both infants and adults with neurological symptoms varying with age of onset. Symptoms include mental retardation, gait problems, cataplexy and ataxia with the most common characteristic being vertical supranuclear gaze palsy. People suffering from Niemann-Pick disease presenting with neuropsychiatric symptoms in combination with delayed onset are often misdiagnosed as other major psychiatric disorders and may thus be prescribed inappropriate treatments. Non-motor signs often precede the first notable clinical motor symptoms in adult Niemann-Pick disease including neuropsychiatric illness in the form of schizophrenia-like psychotic disorders, hallucinations or/and aggressive behaviours. Improved awareness of Niemann-Pick disease and its symptoms among clinicians is essential for better disease detection, since patients diagnosed with chronic psychiatric illness and concomitant neurological impairments managed with antipsychotic drugs may have this disease.
Key words: membranes, glycosphingolipids, neuropsychiatric disorder, gene NPC1, gene NPC2, Niemann-Pick disease type C
ÚVOD
Na základě průkopnických prací Alberta Niemanna a Ludwiga Picka z konce 2. dekády 20. století1 je eponymum Niemannova-Pickova choroba vyhrazeno pro heterogenní skupinu autosomálně recesivně dědičných metabolických onemocnění spojených s poruchou střádání lipidů v lyzosomech, s řadou společných projevů, z nichž k nejčastějším patří hepatosplenomegalie, ukládání sfingomyelinu v retikuloendoteliálních a parenchymatózních tkáních, které doprovází v řadě případů neurologické anebo psychické symptomy.2 Nositel Nobelovy ceny za objevení lyzosomu pro rok 1955 Christian de Duveu3 zahájil pokroky v biochemii, molekulární biologii a patologii lyzosomálního systému, které do dnešních dní dovolily klasifikovat kolem 50 dědičných poruch, souhrnně označovaných jako střádavé poruchy lyzosomu (lysosomal storage disorders - LSD). Společnou charakteristikou je intralyzosomální hromadění směsi látek (kompozit), kterými mohou být substráty nebo produkty nejrůznějších enzymatických systémů, chybně zpracovaných biokonjugátů nebo segregovaných komponent cytoplazmy, které se nacházejí v rozmanitých intracelulárních (nejčastěji intralyzosomálních) lokalizacích, buněčných typech a tkáních.
Největší část LSD je výsledkem deficitní enzymové aktivity způsobené mutací v některém z genů nezbytných pro normální funkci enzymu - tj. mutacemi genu kódujícího enzym samotný, mutací genu protektivního proteinu, mutací proteinových aktivátorů nebo poruchou funkce enzymů endoplazmatického retikula zodpovědných za potranslační modifikaci genového produktu. Další skupina LSD je způsobena poruchou vlastního lyzosomálního systému - tj. mutací genů kódujících komponenty lyzosomální membrány nebo složek asociovaných s lyzosomálním systémem. Počet defektních enzymů a proteinů bez katalytické aktivity popisovaných v literatuře se trvale zvyšuje.4,5
Niemannova-Pickova choroba (NP) v soudobém pojetí zahrnuje dvě odlišné skupiny metabolických poruch: deficit kyselé sfingomyelinázy (výsledek mutace SMPD1 genu zahrnující typy NP A, B a přechodné formy) a alteraci intracelulárního transportu endocytoplazmatického cholesterolu (na podkladě mutace NPC1 a NPC2). Další výzkum reklasifikoval Niemannovu-Pickovu chorobu typu C (NP-C) jako poruchu intracelulárního lipidového obratu zasahujícího více specificky, ale ne pouze endocyto-plazmatický cholesterol. Z klinického hlediska je pro NP-C je charakteristická progredientní neurologická deteriorace vedoucí ke zvýšené morbiditě a předčasnému úmrtí, až u jedné třetiny postižených však v klinickém obraze dominují duševní poruchy. Již v roce 1958 Crocker a Farber6 demonstrovali rozdílnost nástupu prvních příznaků NP-C jakožto široce variabilní kontinuum klinického obrazu, které je v přímé souvislosti s množstvím střádaného sfingomyelinu ve tkáních pacientů. Na základě tohoto zjištění následně navrhl Crocker2 klasifikaci choroby do čtyř podskupin, označovaných A-D: typ A charakterizovaný časnou a těžkou deteriorací funkce centrálního nervového systému (CNS) a masivním střádáním sfingomyelinu ve viscerálních orgánech a v mozku; typ B zahrnující chronický průběh onemocnění s hlavním postižením viscerálních orgánů a relativním ušetřením nervového systému.
NP-C a NP-D byly charakterizovány subakutním poškozením CNS, mírnějším a pomalejším průběhem a slabším postižením viscerálních orgánů (typ D se liší od typu C jen endemicitou výskytu u populace obývající Yarmouth County v Novém Skotsku v Kanadě).
EPIDEMIOLOGIE
Celková kombinovaná prevalence LSD v české populaci se pohybuje okolo 12,25 na 100 000 a je srovnatelná se zeměmi s pokročilými metodami diagnostiky (Nizozemsko 14/100 000, Austrálie 12,9/100 000, Itálie 12,1/100 000). Více než polovinu případů tvoří lipidózy, přibližně jednu čtvrtinu mukopolysacharidózy, dále tzv. NCL (neuronal ceroid lipofuscinosis), mukolipidózy nebo glykoproteinózy7 LSD s nejvyšší prevalencí v ČR při narození je Gaucherova choroba (1,13/100 000 živě narozených). NP-C je s prevalencí 0,91/100 000 (54 pacientů v období 1975 až 2008) druhou nejčastější LSD v České republice. Frekvence přenašečů je asi 1 ze 166.
ETIOLOGIE
NP-C je ubikviterně se vyskytující metabolická porucha způsobená specifickými mutacemi s autosomálně recesivním typem dědičností, které doprovází abnormality v intracelulárním transportu endocytoplazmatického cholesterolu se sekvestrací neesterifikováného cholesterolu a jiných látek v lyzosomech a pozdních endosomech.8 Toto fenotypově velmi homogenní a podobně se projevující střádání vzniká na podkladě dvou rozdílných typů mutací v genech NPC1 a nebo NPC2. Mutace NPC1 zahrnuje většinu případů familiárního výskytu NP-C, včetně původního subtypu D (až 95 % rodin), zatímco mutace NPC2 je poměrně vzácná (bylo celosvětově popsáno asi 30 rodin, odhadem se předpokládá podíl do 4% případů). U zbývajícího 1 % pacientů, ačkoliv biochemicky verifikovaných, nebylo možné popsat konkrétní genetickou mutaci.
Gen NPC1, lokalizovaný na 18. chromosomu, kóduje velký membránový vazebný glykoprotein, jehož dysfunkce vede k intracelulární akumulaci neesterifikovaného cholesterolu a sfingolipidů. Gen NPC2, lokalizovaný na chromosomu 14q24, kóduje malý rozpustný lyzosomální protein s vysokou vazebnou afinitou pro cholesterol. Z důvodů velkého polymorfismu pozorovaných mutací v těchto dvou genech existuje mnoho klinických fenotypu NP-C, které tvoří spíše kontinuum než separátní jednotky. Klinicky není zcela vždy možné přesně rozlišit, zda genotypově došlo k mutaci v NPC1 nebo NPC2 genu.
NEUROPSYCHICKÝ KORELÁT POSTIŽENÍ
Charakteristické pro NP-C je střádání toxických substancí v mozku. Střádán je cholesterol, sfingomyelin, sfingosin, laktosylceramid, glukosylceramid, GM2 gangliosidy, GM3 gangliosidy, přičemž dochází k tvorbě neurofibrilárnich smotků a fosforylovaného tau-proteinu. Především kumulace glykosfingolipidů je zodpovědná za neurologické, behaviorální a kognitivní příznaky, zatímco v játrech dochází především k hromadění cholesterolu, sfingosinu, bismonoacylglycerol-fosfátu a glykosfingolipidů.
Cholesterol je jednou z hlavních komponent buněčné membrány všech buněk v lidském organismu a jeho distribuce a akumulace v těle je velmi pečlivě řízena. Jediným významným mechanismem odstraňování cholesterolu z extrahepatálních tkání je vazba na lipoproteiny a transport do jater k dalšímu zpracování. Potřeba cholesterolu v nervové tkáni mozku je vysoká především v období vývoje a zde hraje hlavní úlohu endogenně syntetizovaný cholesterol. V dospělosti je využíván organismem především cholesterol získaný exogénne. Poškození distribuce cholesterolu během vývoje vede k narušení myelinizace a následné zvýšené pohotovosti neuronů k axonálnímu poškození. Intraneuronální hromadění cholesterolu a porucha schopnosti endogenní či exogénne získaný cholesterol transportovat do distálních částí axonu vede k narušení funkcí buněčné membrány neuronu a porušení schopnosti reagovat na axonální poškození. Axonální struktury se stávají zranitelnými, formují se axonální sféroidní tělíska, dochází k hypomyelinizaci až demyelinizaci v reakci na zvýšené koncentrace kumulovaného cholesterolu. V postižených neuronech dále dochází ke snížení schopnosti arborizace dendritů, což lze přičítat fosforylaci proteinu asociovaného s mikrotubuly Podobné změny nacházíme u pacientů trpících schizofrenií a jinými neuro-degenerativními chorobami.
Hlavními místy poškození mozku jsou dlouhé dráhy v bílé hmotě. Nejvíce zraňovanými místy jsou axony neuronů lokalizované v corpus callosum, mozečku, bazálních gangliích a talamu. Při použití zobrazovacích metod je nacházena bilaterální redukce šedé hmoty v okruzích zapojených v kognitivních a jiných psychických procesech (v hipokampu, talamu, rostrálním oddílu mozečku a v malých oblastech inferoposteriorního kortexu).
KLINICKÁ MANIFESTACE NP-C
Klinické projevy NP-C jsou extrémně heterogenní a různí se především rozdílným věkem manifestace prvních příznaků (od perinatálního období po 6. až 7. dekádu) , dobou přežití (od několika dní po desítky let, nezřídka i 60 let, přestože většina pacientů umírá mezi 10. a 25. rokem věku) a velmi variabilním interindividuálním průběhem.
NP-C se klasicky projevuje jako neuroviscerální onemocnění, kdy jednotlivá orgánová postižení a jejich závažnost jsou na sobě nezávislé. Kromě menšinové části pacientů (úmrtí perinatálně nebo do 6 měsíců věku na jaterní nebo dechové selhání) dojde u většiny pacientů k vývoji chronicky progredientního a často i fatálního neurologického postižení. Až u 45 % nemocných je prvním klinickým příznakem duševní porucha.
U všech věkových kategorií, mimo perinatální formu, je potřeba určit, zda prvotním příznakem je systémové, nebo neurologické postižení. Ukázalo se totiž, že věk při nástupu systémových příznaků není v korelaci se závažností a průběhem neurologického postižení (to se může objevit s latencí desítek let). Na druhé straně ale existuje spojitost mezi věkem nástupu neurologického postižení a dalším průběhem onemocnění a prognózou přežití.
Systémové onemocnění vždy vede k rozvoji neurologických symptomů. Systémová komponenta nemusí být přítomna u 15 % všech pacientů a až u poloviny pacientů s manifestací nemoci v dospělosti. Mimo perinatální období nebývá systémové postižení závažné a bývá dobře snášeno. Splenomegalie (nebo kombinovaná hepato-splenomegalie) mívá kolísavý výskyt s tendencí k ústupu v průběhu času.
Výskyt příznaků dle přehledu literatury9 jsou: mozečková ataxie (75 %), supranukleární paréza vertikálního pohledu (75 %), dysartrie (63 %), kognitivní deficit (61 %), motorický deficit (58 %), splenomegalie (54 %), psychické poruchy (45 %) a dysfagie (37 %).
NEUROLOGICKÉ PROJEVY
NP-C svým patofyziologickým mechanismem v typických případech zasahuje mozeček, bazálni ganglia i kortex a projevuje se především cerebelární ataxií, dysartrií, dysfagií a progresivně postupující demencí. U naprosté většiny pacientů se jako první příznak rozvíjí supranukleární paréza vertikálního pohledu, která vyplývá z postižení mezencefalických struktur. Tento symptom bývá přítomen již v časných stadiích a bývá často, i přes významné snížení rychlosti sakadických očních pohybů, kliniky přehlížen. Ukazuje se, že detekce tohoto symptomu může sloužit jako základní klinický nástroj v diagnostice NP-C. Dalšími častými, avšak ne příliš specifickými příznaky bývají kataplexie s narkolepsií nebo bez narkolepsie (často vyvolaná smíchem), epileptiformní záchvaty (všech typů), ataxie a dystonie.
PSYCHICKÉ PROJEVY
Psychické příznaky jsou přítomny téměř u poloviny pacientů trpících NP-C (uvádí se 45 %). Alespoň u jedné třetiny pacientů se rozvíjejí psychické potíže často několik let před rozvojem jiných příznaků a vyskytují se častěji při pozdějším nástupu nemoci.10
U sledovaných nemocných se rozvinuly jak psychotické fenomény, tak poruchy vnímání, časté byly i behaviorální poruchy vzniklé z "plného zdraví". Pozorované psychotické fenomény byly nej častěji difúzního paranoidního nebo paranoidně-persekučního charakteru, nezřídka se vyskytly formované bludné obsahy. Auditívni a vizuální halucinace bývají méně časté. Heteroagresivní jednání a sebepoškozování jsou většinou přítomny ještě bez přítomnosti neurologického postižení. Dalšími příznaky mohou být poruchy nálady, především čistě depresivního nebo bipolárního charakteru. Pravidelně u postižených dochází k sociálnímu stažení.
Dětské formy jsou typicky spojeny s mentální retardací, epileptiformními záchvaty, ataxií, dystonií a parézou vertikálního pohledu. Adolescentní a adultní formy jsou ve větší míře provázeny neuropsychiatrickým postižením a kognitivním deficitem často směřujícím k plnému rozvoji demence. Z jednotlivých období je třeba zmínit především závažnou pozdně infantilní neurologickou formu (nástup 2-6 let). Hepatosplenomegalie je přítomna po různě dlouhou dobu a současně se ukazuje, že je narušena myelinizace ve frontotemporálním kortexu, přičemž bývá narušena tvorba spojů frontotemporálně a ve striatu. U postižených dochází k opoždění vývoje řeči. Dítě má často problémy s chůzí, padá a je neobratné ve věku mezi třemi a pěti lety (z důvodu mozečkové ataxie). Paréza vertikálního pohledu je ve většině případů přítomna, ale nebývá diagnostikována. U nemocných bývá často popisována ztráta sluchu. Kataplexie se rozvíjí relativně frekventně a v mnoha případech bývá příznakem, pro který je vyhledána lékařská péče. Deteriorace motorických funkcí se postupně prohlubuje a porucha duševního vývoje začíná být více patrná. Signifikantně významná část pacientů rozvine záchvatovité onemocnění, charakter záchvatů může být parciální, generalizovaný nebo jejich kombinace. Epileptiformní projevy reagují na léčbu, vyskytují se však i případy refrakterních záchvatů, při nichž někteří pacienti umírají při status epilepticus nebo na vzniklé komplikace. S prohlubující se ataxií se objevuje dysfagie, dysartrie a rozvíjí se demence. V pozdějším období se objevují pyramidové jevy a roste spasticita, objevují se problémy s polykáním vedoucí k nutnosti zavedení nazogastrické sondy nebo gastrostomie. Postižení se obvykle dožívají 7-12 let. Juvenilní neurologická forma je nejčastější formou NP-C. Středně závažná splenomegalie je častá a může být detekovatelná obvykle již v mladším věku, včetně novorozeneckého období, avšak až v 10 % případů dochází ke spontánní úpravě orgánových příznaků ještě před nástupem neurologické symptomatiky Hlavní diagnostickou svízel představují první nespecifické známky zahrnující školní problémy s obtížemi při psaní a poruchu soustředění (diferenciální diagnóza poruchy pozornosti s hyperaktivitou - ADHD). Tato záměna často vede k nesprávné diagnóze. Téměř jistým příznakem je paréza vertikálního pohledu a jde o první pozorovaný příznak. Dítě se stává neohrabanějším a zhoršují se jeho potíže s učením. Dále bývá přítomna katalepsie s narkolepsií nebo bez narkolepsie, obvykle vyvolaná smíchem. Časem se rozvíjí ataxie s častými pády a neschopností běhu. Onemocnění progreduje velmi individuálně, rozvíjí se dysartrie a dysfagie, objevuje se dystonie s neschopností manipulovat s předměty nebo se samostatně stravovat. Zatímco motorické poškození představuje hlavní problém, snížení intelektových schopností je značně variabilní. Asi polovina pacientů s klasickou formou trpí epileptiformními záchvaty s různým stupněm závažnosti. V pozdějším období se zhoršující se dysartrií pacienti přestávají mluvit, komunikace pokračuje nonverbálně a končí mutismem. V pozdním stadiu se rozvíjejí poruchy hybnosti a spasticita, obtíže s polykáním vedou k nutnosti zavést gastrostomii. Délka dožití je poměrně variabilní a někteří pacienti se dožívají 30 let i déle.
Adultní neurologická forma je charakteristická rozvojem prvních příznaků ve druhé nebo třetí dekádě, výjimečně i v páté a později. Diagnóza NP-C se jeví jako podceňovaná, absence klinicky manifestní splenomegalie je poměrně častým diagnostickým problémem, avšak při sonografickém vyšetření se mohou objevit známky zvětšení sleziny. Neurologické postižení svým průběhem odpovídá vývoji u klasické formy. Epileptické záchvaty jsou poměrně vzácné (u 15 % pacientů). U jedné třetiny nemocných předchází psychická manifestace neurologické symptomatice. Mezi nejčastější psychické příznaky NP-C patří psychózy, zahrnující paranoidní a persekuč-ní bludnou produkci, auditívni a vizuální halucinace, ale i typické intrapsychické halucinace ve formě ozvučení, přenosu nebo odnímání myšlenek. Nástup může být akutní nebo progresivní, eventuálně s relabujícím průběhem. V tomto období může být neurologický nález zcela normální. Další charakteristikou jsou poruchy nálady (především depresivní nebo bipolární), behaviorální problémy spojené s agresivitou nebo sociálním stažením. Byly rovněž popsány případy bipolární afektivní poruchy, obsesivně-kompulzivní poruchy nebo přechodné psychotické poruchy s vizuálními halucinacemi. Nemocní mohou být obecně rozděleni do dvou skupin: u první dominují poruchy motoriky (dystonie, parkinsonismus, chorea), ataxie a dysartrie doprovázené variabilní kognitivní dysfunkcí a druhá skupina trpí spíše dominujícími psychickými poruchami a progredující demencí.
DIAGNOSTIKA NP-C
Klinické podezření
Vyslovit podezření na NP-C se jeví jako relativně snadné při přítomnosti plně vyjádřených typických příznaků (splenomegalie, ataxie, paréza vertikálního pohledu). U juvenilních a infantilních forem se psychické a neurologické příznaky mohou velmi snadno přehlédnout nebo je lze zaměnit za jiná onemocnění anebo mohou zůstat zcela bez povšimnutí. Především význam izolované splenomegalie nebo hepatomegalie jako prvotního symptomu již byl výše zmíněn. Diagnostika je v mnoha případech velmi opožděná. Názor, že vzhledem k omezeným terapeutickým možnostem postačí diagnóza v pozdějších stadiích nemoci, je v dnešní době prvních kauzálních terapeutických metod překonaný.
Při vyslovení podezření na diagnózu NP-C je třeba provést komplexní klinické vyšetření. Neurologické vyšetření má obsahovat testy svalového tonu a testy svalové síly, posouzení svalových reflexů, pohybů (ataxie, dystonie) a test polykání. Psychometrické testy se jeví jako rovněž důležité. Přínosné pro diagnostikuje neurooftalmologické vyšetření.
Vyšetření očních pohybů
Nejčasnějším příznakem bývá porucha sledovacích a sakadických očních pohybů. Odpovídající vyšetření nebývá často provedeno vůbec nebo se mu v kontextu celkového vyšetření nepřikládá patřičná důležitost. Počáteční deficit je v rovině vertikální (pohyb nahoru, dolů nebo oběma směry). Paréza vertikálního pohledu prodloužuje latenci vertikální sakády (obvykle nejprve směrem vzhůru) s postupným zpomalováním sakadických pohybů a finálně kompletní parézou vertikálního pohledu. Pacient sleduje předmět pohybující se vertikálně velmi obtížně a následně ztrácí možnost volního vertikálního očního pohybu zcela. Iniciace či plynulost sakadických i sledovacích horizontálních pohybů očí mohou být také narušeny podobným způsobem.
Vyšetření neurofyziologické a zobrazovací studie
U postižených může být přítomna krátká tranzitorní kataplexie s variabilní závažností postižení od mírných (ztráta svalového tonu v krčním svalstvu spojená s pádem hlavy nebo celého těla, což bývá často zaměňováno s problematikou záchvatů) až po závažné (kolaps celého těla především v reakci na smích) příznaky spojené s narkolepsií.
Významné abnormální nálezy přináší provedení audiogramu nebo kmenových evokovaných potenciálů, a to zejména při postupné ztrátě sluchu z centrálních příčin. Zobrazovací metody (magnetická rezonance nebo komputerová tomografie) nepatří k metodám výtěžným pro stanovení diagnózy NP-C, protože výsledky jsou obvykle spíše normální nebo ukazují jen mírné změny ve smyslu mozečkové a kortikální atrofie. U infantilní formy je možné někdy zachytit změny v obraze magnetické rezonance (dlouhé mozkové dráhy - pyramidové dráhy, corpus callosum, spoje bazálních ganglií aj.) nebo četné T2 hyperechogenitiy v bílé hmotě.
V několika případech byla u nemocných zjištěna periferní neuropatie, potvrzená elektromyografickým vyšetřením.
Vyšetření histologické
V kostní dřeni dochází vlivem narušeného metabolismu lipidů, zejména cholesterolu a sfingomyelinu, k jejich intracelulárnímu hromadění. Tyto změny se někdy projevují přítomností tzv. pěnových buněk, které jsou silně pozitivní při tzv. Filipínském testu. Biopsie kůže, spojivek nebo jater může podpořit diagnózu NP-C. Bez použití Filipínského testu je toto vyšetření zatíženo velkou chybou falešně negativních výsledků.
Vyšetření biochemická
Rutinní laboratorní vyšetření nám většinou neposkytuje příliš mnoho specifických vodítek, výsledky většinou bývají v rozmezí norem. Výjimku tvoří pacienti trpící cholestatickou formou ikteru nebo hypersplenismem. Nízké plazmatické koncentrace HDL-cholesterolu jsou častým, ale ne univerzálním nálezem. Profil plazmatických lipidů je v korelaci se závažností postižení metabolismu cholesterolu. Jako užitečnější se jeví specifičtější testy, např. zvýšená chitotriosidázová aktivita. V leukocytech bývá nacházena normální nebo zvýšená aktivita kyselé sfingomyelinázy (diferenciálně diagnostické vodítko mezi NP-B a atypickou variantou NP-A), na rozdíl od fibroblastů, kde je pozorován deficit tohoto enzymu.
Za vyšetření s nejvyšší senzitivitou a specificitou lze považovat tzv. Filipínský test, který je považován za průkaz narušení intracelulární homeostázy a transportu cholesterolu. Testování se provádí na živých kožních fibroblastech kultivovaných v médiu obohaceném o LDL-cholesterol. Fibroblasty jsou následně fixovány a barveny filipinem, látkou, která vytváří specifické komplexy s neesterifikovaným cholesterolem. Fluorescenční mikroskopií je následně možné prokázat četná perinukleární tělíska. Toto vyšetření provádějí specializovaná centra s dostatečnou zkušeností.
Dle recentní literatury se doporučuje genetické vyšetření u každého případu pacienta s vysloveným podezřením na NP-C. Molekulárně-genetické studie jsou v současné době preferovány především v prenatální diagnostice a jsou prakticky jedinou spolehlivou metodou stanovení přenašečů v pokrevním příbuzenstvu. Navíc, jak bylo zmíněno výše, určení genotypu je v některých případech jedinou možností stanové definitivní diagnózy. Téměř 95% všech pacientů má mutaci v genu NPC1. V malém procentu případů byly prokázány abnormální nálezy pouze v jedné alele, a u několika případů dokonce nebyla nalezena žádná mutace. NP-C jakožto vrozená choroba s auto-somálně recesivní dědičností může být identifikována již prekoncepčně na podkladě rozboru rodinné anamnézy.
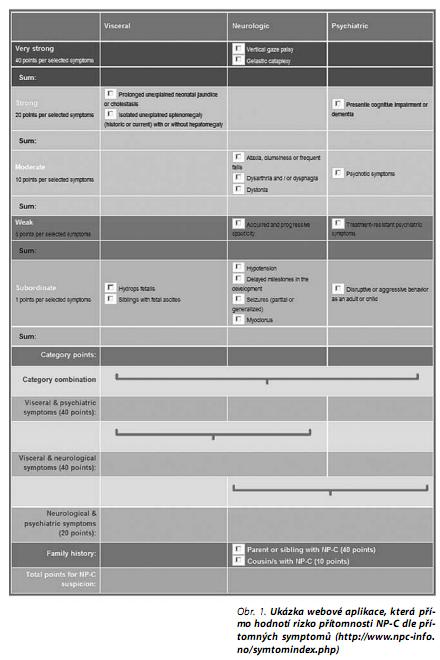
NP-C Symptom Index
NP-C Symptom Index pod byl sestaven na základě četnosti, specificity a senzitivity jednotlivých příznaků u pacientů s projevy NP-C. Vznikl jako pomocné diagnostické vodítko pro zvážení rizika NP-C diagnózy. NP-C Symptom Index byl publikován panelem odborníků z center střádavých onemocnění na Mezinárodním sympoziu Společnosti pro studium dědičných metabolických poruch (SSIEM) v Zenevě v letošním roce. Symptomy jsou rozděleny do tří kategorií - viscerální, neurologické a psychiatrické. Příznaky jsou dále členěny dle jejich diagnostického významu a četnosti výskytu u nemocných NP-C na 5 kategorií.
Přímo dostupná (on-line) mezinárodní verze se nachází na internetové adrese: http://www.npc-info.no/sym-tomindex.php. Lékař zde může označit všechny přítomné symptomy u svého pacienta s podezřením a systém vyhodnotí celkové skóre, jehož hodnota přísluší doporučení dalšího postupu v diagnostice:
- - méně než 40 bodů - nízká pravděpodobnost NP-C;
- - 40 až 69 bodů - nutné sledování a kontaktování NP-C centra;
- - 70 a více bodů - nutné pacienta odeslat k dalšímu vyšetření s podezřením na NP-C ke konzultaci na odborné pracoviště (centrum metabolických vad).
MOŽNOSTI TERAPIE NP-C
Přes rostoucí poznatky soudobé lékařské vědy a jejích pokroků zůstává u vrozených střádavých onemocnění, podobně jako u jiných vrozených onemocnění, kauzální terapie (genové inženýrství) mimo její možnosti - s aspekty etickými, morálními, ekonomickými aj. Podobně jako u jiných infaustních onemocnění platí, že nejdůležitější zásadou je komplexní přístup k pacientovi a všem jeho obtížím. Hlavními pilíři terapie zůstává na prvním místě léčba symptomatická, dále pomoc a podpora postižených rodin ve smyslu genetického poradenství a svépomocných organizací.
SYMPTOMATICKÁ LÉČBA
Záchvatovité projevy všeobecně odpovídají dobře nebo alespoň zčásti na aplikaci antikonvulzivní terapie, a to i v pozdějších stadiích nemoci. Kataplexie je dobře zvládnutelná klomipraminem, protriptylinem nebo modafinilem. Dystonii a tremor se daří u některých pacientů zvládnout především anticholinergně působícími přípravky. Insomnii se daří pozitivně ovlivnit Z-hypnotiky (zopiklonem a zolpidemem) a recentní studie ukazují na možné terapeutické využití melatoninu. V léčbě spasticity a rozvoje kontraktur se uplatňují vhodně zvolené fyzioterapeutické postupy.
Vzhledem k tomu, že za patofyziologický podklad poruchy je považováno hromadění cholesterolu, byly uskutečněny snahy o symptomatické ovlivnění stavu aplikací látek snižujících plazmatické koncentrace cholesterolu vedené snahou minimalizovat jeho intracelulární střádání. Proběhla řada studií s užitím cholestyraminu, lovastatinu, kyseliny nikotinové a dimetylsufoxidu, které snížily množství neesterifikovaného cholesterolu v krvi a játrech s minimem vedlejších efektů, avšak nebyl získán přesvědčivý důkaz o klinickém přínosu této léčby.
Pacienti trpící časným nebo pomalým rozvojem nemoci mohou těžit ze specializovaného vzdělávacího přístupu.
V pozdějších stadiích je samozřejmě namístě správná péče o infekční komplikace (při imobilitě) a potíže s příjmem potravy (nazogastrická sonda, eventuálně gastrostomie).
Symptomatická terapie duševních poruch se v zásadě nijak neliší od terapie primárních psychických onemocnění (podrobněji viz11).
SPECIFICKÁ TERAPIE
V několika případech bylo u pacientů s nálezem mutace genu NPC1 přistoupeno k transplantaci kostní dřeně, avšak neuropsychické potíže se nezmírnily U dětí, u nichž byla transplantace provedena, se nadále zhoršovala kognitivní výkonnost, prohlubovaly se neurologické poruchy, i když došlo k zastavení, nebo dokonce úpravě somatických obtíží (ústup hepatosplenomegalie a snížení infiltrace plic). Podobně i transplantace jater, k níž bylo přistoupeno v několika případech komplikovaných rozvojem cirhózy, nevedla ke zmírnění neuropsychického deficitu.
Jako slibná metoda se testuje transplantace hematopoetických kmenových buněk u pacientů s NPC2 variantou, především protože NPC2 protein je rozpustný prochází fázemi sekrece a vychytávání z a do buněk. Dlouhodobý efekt však dosud není znám, ale dosavadní výsledky získané u jednoho pacienta transplantovaného ve věku 18 měsíců sledovaného do tří let jsou povzbuzující.12
V 90. letech 20. století byla vyslovena hypotéza, že hlavním faktorem zodpovědným za systémová a specifická neuropsychická postižení je intracelulární cholesterol a že cílené snížení vysokých koncentrací lipidů může vést ke zlepšení prognózy onemocnění. V souvislosti s tímto konceptem byla zavedena terapie hypolipidemiky v kombinaci s nízkotučnou dietou. Výsledky ukázaly progresivní zlepšení histologické struktury jater a úpravu jaterních funkcí, ale nevedly ke zvýšení výkonnosti v neuropsychických aspektech.13 Další hypotéza vychází z předpokladu, že k některým neuropatogenním projevům přispívá narušení homeostázy a hromadění glykolipidů v nervových buňkách. Nejprve ve zvířecích modelech a později u lidských dobrovolníků byly provedeny pokusy s inhibitorem glukosylceramidsyntetázy - miglustatem (N-butyl-deoxynojirimycin, NB-DNJ nebo OGT 918). Svým působením zamezuje kompetitivní inhibicí vazbě glukózy na ceramid, a tím snižuje celkové množství vznikajícího glukocerebrosidu. Dochází k redukci množství prekursoru pro dále vznikající glykolipidy a sfingomyeliny, jde tady o tzv. princip deprivace substrátu. Tento přípravek byl následně schválen k terapii mírné až středně závažné formy Gaucherovy choroby typu 1 a nyní i k léčbě NP-C.
Kontrolované klinické pokusy u neurologicky symptomatických pacientů proběhly nejprve u adolescentních a adultních forem (věk nad 12 let) a později i u dětí ve věku 4-12 let. Výsledky dlouhodobého sledování (až 66 měsíců) byly zveřejněny pro dětské, juvenilní i adultní formy.14,15 Na základě posouzení změn v rychlosti horizontálního sakadického očního pohybu, chůze, polykání a kognice došlo ke stabilizaci průběhu až u 72 % případů léčených rok a více. Tento nález vedl komisi EU v lednu 2009 ke schválení indikace miglustatu k léčbě progresivního neurologického deficitu pacientů trpících NP-C. Miglustat tak představuje první specificky účinný lék na Niemannovu-Pickovu choroby typu C.16
ZÁVĚR
NP-C patří mezi střádavá lyzosomální onemocnění s velmi variabilními projevy. Primární manifestace psychickými projevy není výjimkou a tak je nezbytné zvýšit povědomí o této chorobě a její diagnostice tak, aby mohla být včas zahájena její adekvátní léčba. Dosud byla terapie Niemannovy-Pickovy choroby typu C pouze symptomatická, ale v současnosti máme k dispozici terapii, která zasahuje přímo do předpokládaného patogenetického mechanismu onemocnění a snižuje celkové množství vznikajícího glukocerebrosidu. Stabilizace průběhu onemocnění lze dosáhnout u vysokého procenta nemocných.
Autoři článku si dovolují připomenout datum 25. 9. 2011, kdy po delší nemoci zemřel ve veku 72 let prof. MUDr. Milan Elleder, DrSc, bývalý proděkan 1. LF UK a emeritní přednosta Ústavu dědičných metabolických poruch 1. lékařské fakulty Univerzity Karlovy a Všeobecné fakultní nemocnice v Praze. Profesor Elleder se systematicky zabýval studiem patogenetických mechanismů u lyzosomálních onemocnění a v problematice Niemannovy-Pickovy choroby a dalších střádavých onemocnění byl mezinárodně uznávanou autoritou, která stála při klasifikaci těchto nemocí.
LITERATURA
- 1. Vanier, MT. Niemann-Pick disease type C. Orphanet Journal of Rare Diseases 2010; 5: 16.
- 2. Crocker AC. The cerebral defect in Tay-Sachs disease and Niemann-Pick disease. J Neurochem 1961; 7: 69-80.
- 3. De Duve C, Pressman BC, Gianetto R, Wattiaux R, Appelmans F. Tissue fractionation studies: 6. Intracellular distribution patterns of enzymes in rat-liver tissue. Biochem J1955; 60 (4): 604-617.
- 4. Balreira A, Caspar P, Caiola D et al. A nonsense mutatuion in the LIMP-2 gene associated with progressive myoclonic epilepsy and nephrotic syndrome. Hum Mol Genet 2008; 17 (14): 2238-2243.
- 5. Berkovic SF, Dibbens LM, Oshlack A et al. Array-based gene discovery with three unrelated subjects shows SCARB2/LIMP-2 deficiency causes myoclonus epilepsy and glomerulosclerosis. Am J Hum Genet 2008; 82 (3): 673-684.
- 6. Crocker AC, Farber S. Niemann-Pick disease: a review of eighteen patients. Medicine (Baltimore) 1958; 37: 1-95.
- 7. Poupětová H, Ledvinová J, Berná L et al. The birth prevalence of lysosomal storage disorders in the Czech Republic: comparison with data in different populations. J Inherit Metab Dis 2010; 33 (4): 387-396.
- 8. Peake KB, Vance JE. Defective cholesterol trafficking in Niemann-Pick Cdeficient cells. FEBS Lett 2010; 584 (13): 2731-2739.
- 9. Sevin M, Lesca G, Baumann N et al. The adult form of Niemann-Pick disease type C Brain 2007; 130: 120-133.
- 10. Yanjanin NM, Velez JI, Gropman A et al. Linear clinical progression, independent of age of onset, in Niemann-Pick disease, type C. Am J Med Genet B Neuropsychiatr Genet 2009; 153B: 132-140.
- 11. Wraith JE et al. Recommendations on the diagnosis and management of Niemann-Pick disease type C. Mol Genet Metab 2009; 98: 152-165.
- 12. Bonney DK, O'Meara A, Shabani A et al. Successful allogeneic bone marrow transplant for Niemann-Pick disease type C2 is likely to be associated with a severe "graft versus substrate" effect. J Inherit Metab Dis 2010 in press (DOI 10.1007/sl0545-010-9060-3).
- 13. Schiffmann R. Niemann-Pick disease type C. From bench to bedside. JAMA 1996; 276: 561-564.
- 14. Patterson MC, Vecchio D, Jacklin E et al. Long-term miglustat therapy in children with Niemann-Pick disease type C. J Child Neurol 2010; 25: 300-305.
- 15. Wraith JE, Vecchio D, Jacklin E et al. Miglustat in adult and juvenile patients with Niemann-Pick disease type C: long-term data from a clinical trial. Mol Genet Metab 2010; 99: 351-357.
- 16. Wraith JE, Imrie J. New therapies in the management of Niemann-Pick type C disease: clinical utility of miglustat. TherClin Risk Manag 2009; 5:877-887.